Neural G0: a quiescent-like state found in neuroepithelial-derived cells and glioma
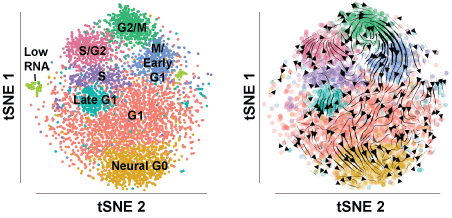
Table of Contents
- Abstract
- Data and code availability
- Instructions to set up data and code for recreating analyses
- Docker container
- Analyses
- Identification of cell cycle phases
- Resolving the flow of cells through the cell cycle using RNA velocity
- Prepare data for building classifier
- Build classifier: 100-fold cross-validation
- Sensitivity analysis
- Whitfield et al., 2002 gold-standard classification
- Classify human scRNA-seq datasets
- Classify mouse scRNA-seq datasets
- Classify glioma scRNA-seq datasets
- ccAF classifier
- Contact
- Citation
Abstract
In depth knowledge of the cellular states associated with normal and disease tissue homeostasis is critical for understanding disease etiology and uncovering therapeutic opportunities. Here, we used single cell RNA-seq to survey the cellular states of neuroepithelial-derived cells in cortical and neurogenic regions of developing and adult mammalian brain to compare with 38,474 cells obtained from 59 human gliomas, as well as pluripotent ESCs, endothelial cells, CD45+ immune cells, and non-CNS cancers. This analysis suggests that a significant portion of neuroepithelial-derived stem and progenitor cells and glioma cells that are not in G2/M or S phase exist in two states: G1 or Neural G0, defined by expression of certain neuro-developmental genes. In gliomas, higher overall Neural G0 gene expression is significantly associated with less aggressive gliomas, IDH1 mutation, and extended patient survival, while also anti-correlated with cell cycle gene expression. Knockout of genes associated with the Hippo/Yap and p53 pathways diminished Neural G0 in vitro, resulting in faster G1 transit, down-regulation of quiescence-associated markers, and loss of Neural G0 gene expression. Thus, Neural G0 is a dynamic cellular state required for indolent cell cycles in neural-specified stem and progenitors poised for cell division. As a result, Neural G0 occupancy may be an important determinant of glioma tumor progression.
Data and code availability
- All datafiles needed to run this code can be found on figshare.
- The analysis software and scripts are available on github.
Instructions to setup data and code for recreating analyses
In order to run the software and scripts you will need to set up a specific directory structure and download all of the data and scripts. Here are the instructions to set things up:
- Clone the U5_hNSC_Neural_G0 repository
git clone https://github.com/plaisier-lab/U5_hNSC_Neural_G0.git - Make a “data” folder inside the U5_hNSC_Neural_G0 folder
cd U5_hNSC_Neural_G0 mkdir data cd data - Download (and unzip for zip files) all files from figshare:
- U5_hNSC.zip (needs to be unzipped) - contains all the U5-hNSC scRNA-seq datasets as output from cellranger.
- ccAF_1536_smaller.pkl (does not need to be unzipped) - the ccAF ACTINN loadings for classification of cell cycle phases for cells or transcriptome profiles.
- geneConversions.zip (needs to be unzipped) - helpful gene ID conversion files.
- forClassification.zip (needs to be unzipped) - loom data files that were classified using ccAF.
- ssGSEA.GBM.classification.zip (needs to be unzipped) - subtype classification results for all glioma datasets.
- Whitfield.zip (needs to be unzipped) - Whitfield et al., 2002 gold-standard dataset for validation of S & M phases.
- cellcycle_int_integrated.loom (does not need to be unzipped) - U5 hNSC data as a loom file that was used to build the ccAF classifier.
wget https://ndownloader.figshare.com/files/24130952 -O U5_hNSC.zip wget https://ndownloader.figshare.com/files/24128372 -O ccAF_1536_smaller.pkl wget https://ndownloader.figshare.com/files/24130958 -O geneConversions.zip wget https://ndownloader.figshare.com/files/24130970 -O forClassification.zip wget https://ndownloader.figshare.com/files/24128375 -O ssGSEA_GBM_classification.zip wget https://ndownloader.figshare.com/files/24128369 -O cellcycle_int_integrated.loom wget https://ndownloader.figshare.com/files/24130961 -O Whitfield.zip unzip U5_hNSC.zip geneConversions.zip forClassification.zip ssGSEA.GBM.classification.zip Whitfield.zip
Directory structure
After downloading and unzipping the files, the directory structure should look like this:
.
+-- U5_hNSC_Neural_G0
| +-- actinn.py
| +-- calculatingErrors_CV.py
| +-- calculatingErrors_Whitfield.py
| +-- classifiersV3.py
| +-- classifyPrimaryCells_gliomas.py
| +-- classifyPrimaryCells_homoSapeins.py
| +-- classifyPrimaryCells_musMusculus.py
| +-- converting_to_loom.R
| +-- cvClassification_FullAnalysis.py
| +-- data
| | +-- ccAF_1536_smaller.pkl
| | +-- cellcycle_int_integrated.loom
| | +-- forClassification
| | | +-- gliomas
| | | | +-- Bhaduri.loom
| | | | +-- GSE102130.loom
| | | | +-- GSE131928_10X.loom
| | | | +-- GSE131928_Smartseq2.loom
| | | | +-- GSE139448.loom
| | | | +-- GSE70630.loom
| | | | +-- GSE84465.loom
| | | | +-- GSE84465_all.loom
| | | | +-- GSE89567.loom
| | | +-- GSE103322.loom
| | | +-- GSE67833.loom
| | | +-- HEK293T.loom
| | | +-- Nowakowski_norm.loom
| | | +-- PRJNA324289.loom
| | +-- geneConversions
| | | +-- ensembl_entrez.csv
| | | +-- hgnc_geneSymbols.txt
| | | +-- hgnc_geneSymbols_ensmbl.txt
| | | +-- human_hgnc_mouse_mgi.csv
| | | +-- mart_export.txt
| | +-- highlyVarGenes_WT_sgTAOK1_1584.csv
| | +-- ssGSEA.GBM.classification
| | | +-- p_result_Bhaduri_2019.gct.txt
| | | +-- p_result_GSE102130.gct.txt
| | | +-- p_result_GSE131928_GSM3828672.gct.txt
| | | +-- p_result_GSE131928_GSM3828673_1.gct.txt
| | | +-- p_result_GSE131928_GSM3828673_2.gct.txt
| | | +-- p_result_GSE139448.gct.txt
| | | +-- p_result_GSE70630.gct.txt
| | | +-- p_result_GSE84465.gct.txt
| | | +-- p_result_GSE89567.gct.txt
| | | +-- res_All_GSE131928.csv
| | +-- U5_hNSC
| | | +-- sgTAOK1
| | | | +-- filtered_gene_bc_matrices
| | | | | +-- hg19
| | | | | | +-- barcodes.tsv
| | | | | | +-- genes.tsv
| | | | | | +-- matrix.mtx
| | | +-- WT
| | | | +-- filtered_gene_bc_matrices
| | | | | +-- hg19
| | | | | | +-- barcodes.tsv
| | | | | | +-- genes.tsv
| | | | | | +-- matrix.mtx
| | | | | +-- tsne_cell_embeddings_Perplexity_26.csv
| | | | | +-- U5_all_v3.loom
| | | | | +-- U5_velocyto.loom
| | | +-- WT_CDTplus
| | | | +-- filtered_gene_bc_matrices
| | | | | +-- hg19
| | | | | | +-- barcodes.tsv
| | | | | | +-- genes.tsv
| | | | | | +-- matrix.mtx
| | +-- Whitfield
| | | +-- data
| | | | +-- whitfield_dataPlusScores_6_30_2020_SHAKE.T_1134.csv
| | | | +-- whitfield_dataPlusScores_6_30_2020_SHAKE_1134.csv
| | | | +-- whitfield_dataPlusScores_6_30_2020_TN.T_1134.csv
| | | | +-- whitfield_dataPlusScores_6_30_2020_TN_1134.csv
| | | | +-- whitfield_dataPlusScores_6_30_2020_TT1.T_1134.csv
| | | | +-- whitfield_dataPlusScores_6_30_2020_TT1_1134.csv
| | | | +-- whitfield_dataPlusScores_6_30_2020_TT2.T_1134.csv
| | | | +-- whitfield_dataPlusScores_6_30_2020_TT2_1134.csv
| | | | +-- whitfield_dataPlusScores_6_30_2020_TT3.T_1134.csv
| | | | +-- whitfield_dataPlusScores_6_30_2020_TT3_1134.csv
| | | +-- markergenes_ForPlotting.csv
| | | +-- metaInformation.csv
| +-- LICENSE
| +-- makeDatasetsForClassification.R
| +-- plotNowakowski.py
| +-- plottingClassifiers.py
| +-- README.md
| +-- results
| +-- sensitivityAnalysis_plot.py
| +-- sensitivityAnalysis_run.py
| +-- ssgse.GBM.classification
| +-- U5_hNSC_scRNA_seq_Analysis.R
| +-- Whitfield_classification_ACTINN_analysis.py
Now, make a results directory to hold the output from analysis scripts:
mkdir results
Docker container
We facilitate the use of our code and data by providing a Docker Hub container cplaisier/scrna_seq_velocity, which has all the dependencies and libraries to run the scripts. To see how the Docker container is configured, please refer to the Dockerfile. Please install Docker and then from the command line, run:
docker pull cplaisier/scrna_seq_velocity
Then run the Docker container using the following command (replace
docker run -it -v '<the directory holding U5_hNSC_Neural_G0>:/files' cplaisier/scrna_seq_velocity
This will start the Docker container in interactive mode and will leave you at a command prompt. You will then want to change directory to ‘/files/U5_hNSC_Neural_G0’ (note the name of the container ef02b3a45938 will likely be different for your instance):
root@ef02b3a45938:/tmp/samtools-1.10# cd /files/U5_hNSC_Neural_G0
root@ef02b3a45938:/files/U5_hNSC_Neural_G0#
If you are able to change to the ‘U5_hNSC_Neural_G0 directory’ you should be ready to move on to the Analyses below.
Analyses
The order of analyses in this study and the details of each analysis are described below:
1. Identification of cell cycle phases
NOTE! This code requires an extra installation step to revert the R package Seurat to V2.3.4. To install Seurat V2.3.4, please run the following command:
root@ef02b3a45938:/files/U5_hNSC_Neural_G0# R > source("https://z.umn.edu/archived-seurat")Once this portion of the analysis is completed, please close the Docker instance by typing ‘exit’ into the console until you return to your base operating system. Then restart the Docker instance as described above.
Using scRNA-seq we profiled 5,973 actively dividing U5-hNSCs (Bressan et al, 2017) to identify the single-cell gene expression states corresponding to the cell cycle phases with a focus on G0/G1 subpopulations. This will take in the scRNA-seq data from the ‘data/U5_hNSC’ directory where the 10X cellranger outputs for the U5-hNSCs are stored.
root@ef02b3a45938:/files/U5_hNSC_Neural_G0# Rscript U5_hNSC_scRNA_seq_Analysis.R
This script will output:
- results/tsne_cell_embeddings_Perplexity_26.csv - TSNE embeddings for use in later plots.
- results/eightClusters_WT_sgTAOK1.csv - marker genes that discriminate between U5-hNSC cell cycle clusters.
- results/TSNE_perplexity_26.pdf - TSNE plot for U5-hNSC WT.
- results/cellCycleNetwork.pdf - network that shows how each cell cycle cluster connects to the other clusters.
- Three hypergeometric p-values for overlaps with YAP target genes which are printed out to the console.
2. Resolving the flow of cells through the cell cycle using RNA velocity
We added directionality to the edges using RNA velocity which computes the ratio of unspliced to spliced transcripts and infers the likely trajectory of cells through a two-dimensional single cell embedding, e.g. tSNE. The RNA velocity trajectories delineate the cell cycle in the expected orientation. First, we use velocyto to realign the transcriptome and tabulate spliced and unspliced transcript counts for each gene (we provide the result of this and not the raw data; the genome build from 10X is very large genome build used from cellranger):
root@ef02b3a45938:/files/U5_hNSC_Neural_G0# velocyto run10x -m hg19_rmsk.gtf WT genes.gtf
This analysis will output:
- data/U5_hNSC/WT/U5_velocyto.loom - a loom file with the matrix of spliced and unspliced reads for each gene.
Then, we use scvelo to take in the unspliced and spliced transcript counts and compute the RNA velocities and stream lines.
root@ef02b3a45938:/files/U5_hNSC_Neural_G0# python3 scvelo_analysis.py
This script will output:
- results/ccAdata_velocity_stream_tsne.png - tSNE embeddings with RNA velocity stream lines.
3. Prepare data for building classifier
We facilitate further analysis in Python by converting the WT U5-hNSC data into a loom file:
root@ef02b3a45938:/files/U5_hNSC_Neural_G0# Rscript converting_to_loom.R
This script will output:
- results/highlyVarGenes_WT_sgTAOK1_1584.csv - list of the overlapping top 2,000 highly variable genes from UT and sgTAOK1.
- results/cellcycle_int_integrated.loom - loom file used to construct the ccAF classifier.
4. Build classifier: 100-fold cross-validation
We used the hNSC scRNA-seq data to build a cell cycle classifier. We tested four different methods which were previously found to be useful for building classifiers from scRNA-seq profiles:
- Support Vector Machine with rejection (SVMrej)
- Random Forest (RF)
- scRNA-seq optimized K-Nearest Neighbors (KNN)
- scRNA-seq optimized ACTINN Neural Network (NN) method.
We selected the 1,536 most highly variable genes in the U5-hNSC scRNA-seq profiles as the training dataset for the classifier. We then used 100-fold cross-validation (CV) to determine the best method:
root@ef02b3a45938:/files/U5_hNSC_Neural_G0# python3 cvClassification_FullAnalysis.py
This script will output two files for each method tested:
- results/<method>/ccAF_CV_results.csv - The true labels and predicted labels for the test sets from each cross-validation iteration. The <method> will be replaced with the name of the method: SVMrej, RF, KNN, and ACTINN.
- results/<method>/CV_classification_report.csv - Classification reports that have F1 scores and other metrics for classifier quality control. The <method> will be replaced with the name of the method: SVMrej, RF, KNN, and ACTINN.
This script will calculate the errors for each method:
root@ef02b3a45938:/files/U5_hNSC_Neural_G0# python3 calculatingErrors_CV.py
This script will output:
- results/errors_cross_validation.csv - The error rate based on the 100-fold CV.
This script makes box plots of the F1 quality control metrics:
root@ef02b3a45938:/files/U5_hNSC_Neural_G0# python3 plottingClassifiers.py
This script will output:
- results/cell_cycle_states_CV_stats.csv - Mean of stats for each classifier method and cell cycle state.
- results/CV_plots/F1_scores_for_each_state_per_classifier_1536.pdf - A boxplot of each classifier method stratified by cell cycle state.
- results/CV_plots/F1_scores_for_each_state_per_classifier_1536_2.pdf - A boxplot of each cell cycle state stratified by classifier method.
5. Sensitivity analysis
A major issue in scRNA-seq studies is that the number of genes detected is heavily dependent upon sequencing depth and missing gene information is commonplace in scRNA-seq studies. Therefore, we conducted a sensitivity analysis to determine the effect of randomly removing an increasing percentage of genes.
root@ef02b3a45938:/files/U5_hNSC_Neural_G0# python3 sensitivityAnalysis.py
This script will output:
- results/SensitivityAnalysis/ccAF_CV_sensitivity_analysis.csv - The true labels and predicted labels for the test sets from each cross-validation iteration.
- results/SensitivityAnalysis/ccAF_CV_sensitivity_analysis_boxplot.pdf - Boxplot showing how increasing amounts of missing genes affects the error rate of the ccAF classifier.
6. Whitfield et al., 2002 gold-standard classification
We validated S and M phase classifications by applying the ccAF classifier to a gold standard cell-cycle synchronized time-series dataset from HeLa cells with simultaneous characterization of transcriptome profiles and experimental determination of whether the cells were in S or M phase at each time point:
Whitfield et al., 2002 - gold-standard dataset of 1,134 most cyclic genes was used to validate S & M phases from ccAF (http://genome-www.stanford.edu/Human-CellCycle/HeLa/).
root@ef02b3a45938:/files/U5_hNSC_Neural_G0# python3 Whitfield_classification_ACTINN_analysis.py
This script will output:
- results/Whitfield/ACTINN_results_Whitfield_1134_quantile.csv - The true labels and predicted labels for the test sets from each cross-validation iteration.
- results/Whitfield/ACTINN_classification_report_Whitfield_1134_quantile.csv - Classification reports that have F1 scores and other metrics for classifier quality control.
- results/Whitfield/plots/F1_scores_for_each_state_Whitfield_1134_quantile.pdf - A boxplot of each classifier method stratified by cell cycle state.
- results/Whitfield/plots/F1_scores_for_each_state_Whitfield_1134_quantile_2.pdf - A boxplot of each cell cycle state stratified by classifier method.
- results/Whitfield/plots/heatmapsOfWhitfield_1134_quantile.csv - Recreating Figure 2 from Whitfield et al., 2002 with ccAF cell cycle phase classification.
7. Classify human scRNA-seq datasets
We classified three human scRNA-seq studies:
- Noakowski et al., 2017 - allowed us to investigate how Neural G0 might arise during mammalian development by applying the ccAF to data from the developing human telencephalon.
- HEK293T - 3,468 HEK293T cells from a barnyard assay conducted by 10X.
- Puram et al., 2017 - scRNA-seq from head and neck squamous cell carcinoma (HNSCC) tumors (GSE103322). Run this command to classify the cells from these human scRNA-seq studies:
root@ef02b3a45938:/files/U5_hNSC_Neural_G0# python3 classifyPrimaryCells_homoSapiens.py
This script will output two files for each study:
- results/ccAF_results_*.csv - a table where the cells are rows and the columns are meta-information about the cells. ‘Predictions’ are the predicted ccAF labels. The asterisk will be replaced with the name of the study: Nowakowski_norm, HEK293T, and GSE103322.
- results/table1_*.csv - a confusion matrix of counts for each study. The asterisk will be replaced with the name of the study: Nowakowski_norm, HEK293T, and GSE103322.
The Nowakowski et al., 2017 results were then plotted to be added to Figure 2:
root@ef02b3a45938:/files/U5_hNSC_Neural_G0# python3 plotNowakowski.py
This script will output two files for each study:
- results/results/Nowakowski_ccAF_plot_Refined.pdf - a stacked barplot that shows how the cells form each cell type in Nowakowski et al., 2017 are classified by ccAF.
8. Classify mouse scRNA-seq datasets
We classified two mouse scRNA-seq studies:
- Llorens-Bobadilla et al., 2015 - defined quiescent neural stem cell (qNSC) and active (aNSC) subpopulations from adult mouse subventricular zone (GSE67833).
- Dulken et al., 2017 - perform single cell transcriptomics on neural stem cells (NSCs) from adult mice (PRJNA324289). Run this command to classify the cells from these mouse scRNA-seq studies:
root@ef02b3a45938:/files/U5_hNSC_Neural_G0# python3 classifyPrimaryCells_musMusculus.py
This script will output two files for each study:
- results/ccAF_results_*.csv - a table where the cells are rows and the columns are meta-information about the cells. ‘Predictions’ are the predicted ccAF labels. The asterisk will be replaced with the name of the study: GSE67833 and PRJNA324289.
- results/table1_*.csv - a confusion matrix of counts for each study. The asterisk will be replaced with the name of the study: GSE67833 and PRJNA324289.
9. Classify glioma scRNA-seq datasets
We classified seven human scRNA-seq studies:
- Tirosh et al., 2016 - grade II oligodendrogliomas that are IDH1 mutant (GSE70630).
- Venteicher et al., 2017 - grade III astrocytomas that are IDH1 mutant (GSE89567).
- Darmanis et al., 2017 - grade IV glioblastomas that are IDH wild-type (GSE84465).
- Neftel et al., 2019 - grade IV glioblastomas that are IDH wild-type (GSE131928).
- Bhaduri et al., 2020 - grade IV glioblastomas that are IDH wild-type (PRJNA579593).
- Wang et al., 2020 - grade IV glioblastomas that are IDH wild-type (GSE139448).
- Filbin et al., 2018 - diffuse midline glioma with H3K27M (GSE102130)
Run this command to classify the cells from these glioma scRNA-seq studies:
root@ef02b3a45938:/files/U5_hNSC_Neural_G0# python3 classifyPrimaryCells_gliomas.py
This script will output two files for each study:
- results/ccAF_results_*.csv - a table where the cells are rows and the columns are meta-information about the cells. ‘Predictions’ are the predicted ccAF labels. The asterisk will be replaced with the name of the study: GSE70630, GSE89567, GSE854465_all, GSE131928_10X, GSE131928_Smartseq2, Bhaduri, GSE139448, GSE102130.
- results/table1_*.csv - a confusion matrix of counts for each study. The asterisk will be replaced with the name of the study: GSE70630, GSE89567, GSE854465_all, GSE131928_10X, GSE131928_Smartseq2, Bhaduri, GSE139448, GSE102130.
ccAF classifier
The ccAF classifier is currently available for download, installation, and execution as either:
- PyPi Python package:
- Install using:
pip install ccAF
- Install using:
- Docker container:
- Pull using:
docker pull cplaisier/ccaf-Run using:
docker run -it -v '<path to files to classify>:/files' cplaisier/ccafMore detailed instructions on using the ccAF classifier can be found with the github repository: https://github.com/plaisier-lab/ccAF
Contact
For issues or comments please contact: Samantha O’Connor or Chris Plaisier
Citation
Neural G0: a quiescent-like state found in neuroepithelial-derived cells and glioma. Samantha A. O’Connor, Heather M. Feldman, Chad M. Toledo, Sonali Arora, Pia Hoellerbauer, Philip Corrin, Lucas Carter, Megan Kufeld, Hamid Bolouri, Ryan Basom, Jeffrey Delrow, José L. McFaline-Figueroa, Cole Trapnell, Steven M. Pollard, Anoop Patel, Patrick J. Paddison, Christopher L. Plaisier. bioRxiv 446344; doi: https://doi.org/10.1101/446344